East Asian Arch Psychiatry 2024;34:141-6 | https://doi.org/10.12809/eaap2435
PERSPECTIVE
Abstract
This review aims to determine the role of S-adenosylmethionine (SAMe) supplementation as an alternative therapeutic option, particularly for individuals with inadequate responses to conventional antidepressive treatments. The effects of SAMe on depression are analysed through its role in modulating neurotransmitter metabolism, reducing neuroinflammation, enhancing neuroplasticity, and regulating gene expression. These mechanisms may contribute to the efficacy of SAMe in treating depression, particularly in treatment-resistant cases. The review also addresses SAMe’s potential use in managing other psychiatric disorders and neurological diseases.
Sonali Labhade, Dr DY Patil Institute of Pharmaceutical Sciences and Research, Pimpri, Pune, India
Ritesh Bhole, Dr DY Patil Institute of Pharmaceutical Sciences and Research, Pimpri, Pune, India; Dr DY Patil Dental College and Hospital, Dr DY Patil Vidyapeeth, Pimpri, Pune, India
Smita Jain, Central University of Rajasthan, India
Address for correspondence: Dr Sonali Labhade, Dr DY Patil Institute of Pharmaceutical Sciences and Research, Pimpri, Pune, India. Email: sonali.labhade@dypvp.edu.in
Submitted: 2 August 2024; Accepted: 18 October 2024
Depression is characterised by enduring feelings of sadness, diminished interest or enjoyment, and impaired daily functioning. Despite availability of antidepressive medications, a proportion of patients still fail to attain adequate symptom relief or experience intolerable adverse effects.1 These patients with treatment-resistant depression require alternative treatment options. Depression is a multifaceted psychiatric condition marked by disturbances in mood, cognition, and behaviour. However, conventional antidepressive medications primarily target monoaminergic neurotransmission only. S-adenosylmethionine (SAMe), a naturally occurring compound involved in various neurobiological processes of mood regulation, presents a promising treatment option.2,3
We reviewed meta-analyses, randomised controlled trials, longitudinal studies, and combination therapy studies to elucidate SAMe’s mechanisms of action and its potential efficacy in managing depression. We also explored SAMe supplementation as an adjunctive therapy for individuals with treatment-resistant depression, its applications in other psychiatric disorders, and its effect on neurological diseases.
S-adenosylmethionine
SAMe is an endogenous compound involved in methylation reactions, neurotransmitter synthesis, and antioxidant defence mechanisms. SAMe serves as a methyl donor in transmethylation reactions, facilitating the synthesis of neurotransmitters such as serotonin, dopamine, and norepinephrine. Additionally, SAMe modulates neuroinflammatory pathways, enhances neuroplasticity, and regulates gene expression implicated in mood regulation. These mechanisms underlie the potential antidepressive effects of SAMe.4
Neurotransmitter metabolism
SAMe has been used for treating a variety of conditions, including depression, and is marketed as a mood and emotional wellbeing enhancer.5 The synthesis of SAMe from L-methionine occurs through a metabolic pathway called the one-carbon cycle. This partially depends on sufficient amounts of the vitamins folate and B12, and deficiency of these vitamins is associated with a higher risk of depression.6 For instance, 10% to 30% of individuals with depression have low folate concentrations and react less favourably to antidepressants. Conversion of vitamin B12 to methylcobalamin plays a crucial role in the synthesis of various neurotransmitters within the central nervous system; therefore, a lack of vitamin B12 might lead to an earlier onset of depression. Decreased levels of SAMe may be a common mediator, and therefore correcting deficiencies in folate and vitamin B12 can potentially alleviate symptoms of depression and improve the effectiveness of antidepressive medications, probably by increasing the concentration of SAMe.7 Individuals with depression have reduced quantities of SAMe in their cerebrospinal fluid; an increase in plasma SAMe concentrations is proportionally associated with an improvement in depressed symptoms.8 Individuals with depression and schizophrenia exhibit reduced activity of the enzyme methionine adenosyltransferase, which is involved in SAMe synthesis; however, the activity level is high in patients with manic episodes. Thus, SAMe may act as a mood-enhancing substance.
Modulation of neuroinflammation
Inflammation of the brain is associated with the onset of chronic conditions such as depression,9 schizophrenia,10 and Alzheimer disease (AD).11 SAMe reduces cytokine and interleukin levels in lipopolysaccharide- stimulated monocytes and macrophages, acting as an anti-inflammatory agent.12,13 SAMe’s pharmacological inhibition of inflammation mediators involves modification of the methylation capability and inhibition of the binding of trimethylated H3K4 to the tumour necrosis factor-α promoter, which increases methylthioadenosine and S- adenosylhomocysteine levels.14 Furthermore, administration of SAMe decreases phosphodiesterase 4B expression by enhancing the attachment of transcriptionally restrictive trimethylated H3K9 to its promoter region, consequently impeding tumour necrosis factor-α generation mediated by cyclic adenosine monophosphate.13 There is a decrease in the DNA methylation status of inflammatory genes in human macrophages following SAMe treatment.15,16 Therefore, the anti-inflammatory efficacy of SAMe, via methylation regulation, may have effects on chronic inflammatory conditions.17-19
Enhancement of neuroplasticity
Neuroplasticity is the capacity of the brain to adapt and reorganise itself in response to various stimuli; it is crucial for regulating mood and responding to antidepressive treatments. SAMe enhances neuroplasticity by promoting synaptic neurotransmission, neurogenesis, and dendritic spine density in key brain regions implicated in depression such as the prefrontal cortex and hippocampus. By facilitating structural and functional neuronal changes, SAMe fosters adaptive responses to stress and enhances resilience against depressive pathology.20
Regulation of gene expression
DNA methylation is involved in the control of gene expression and the development and maintenance of cellular identity. DNA methylation can be disturbed by a variety of illnesses; gene expression can remain adaptable throughout time in response to variables that are either developmental or environmental.21-23 Many DNA methylation markers have been implicated in the dynamics of gene regulation in the central nervous system (Table and Figure).24-27

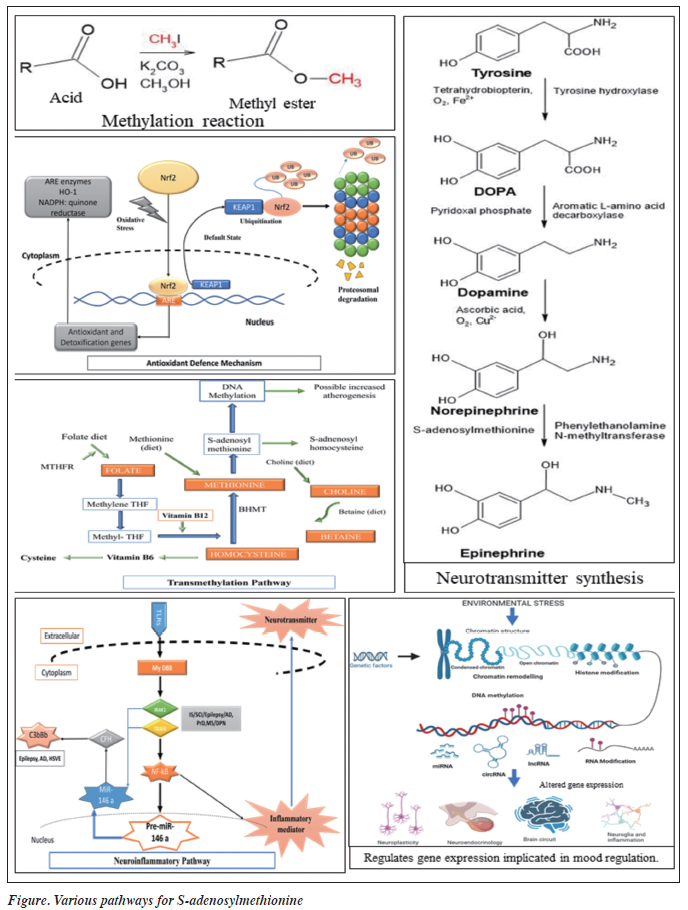
Clinical evidence
There is robust clinical evidence supporting the efficiency of SAMe supplementation in alleviating symptoms of depression, particularly in patients who have not responded optimally to standard antidepressive therapy.8,28-31
SAMe versus placebo as adjunctive treatment
Oral administration of SAMe at dosages of 800 to 1600 mg/day resulted in significant reductions in depressive symptoms.8 In a 6-week trial comparing SAMe (800 mg/ twice daily) with placebo, the Hamilton Depression Rating Scale score response rate (36.1% vs 17.6%) and remission rate (25.8% vs 11.7%) were significantly higher among patients treated with adjunctive SAMe than with adjunctive placebo.19
SAMe versus imipramine or escitalopram as adjunctive treatment
SAMe’s efficacy as an adjunctive therapy for depression stems from its role as a methyl donor in vital biochemical reactions, including the synthesis and metabolism of key neurotransmitters such as serotonin, dopamine, and norepinephrine. SAMe promotes mood regulation and alleviates depressive symptoms. SAMe has been shown to reduce inflammation and oxidative stress, both of which are central to the pathophysiology of depression. SAMe can enhance the antidepressant effects of existing treatments, particularly in patients with major depressive disorder who have not responded adequately to selective serotonin reuptake inhibitors alone.
SAMe accelerates the onset of imipramine’s antidepressive effects in patients with depression, potentially due to their combined action on serotonin 1A (5-HT1A) receptors.25 Compared with saline, imipramine alone upregulated the 5-HT1A receptor levels in the frontal cortex but failed to induce an antidepressive effect, whereas SAMe alone or in combination with imipramine did not affect 5-HT1A receptor levels in the frontal cortex but showed antidepressive effects. Long-term treatments with SAMe, imipramine, or their combination resulted in both antidepressive effects and upregulation of 5-HT1A receptor levels in the hippocampus. Interestingly, SAMe inhibited the upregulation of 5-HT1A receptors in the frontal cortex caused by imipramine. This interaction may contribute to the antidepressive effect of imipramine.
SAMe, escitalopram, and placebo resulted in significant improvement in depression symptoms, as measured by the Hamilton Depression Rating Scale, from baseline to week 12.26 The effect size for SAMe versus placebo was moderate to large (d=0.74). SAMe was superior to placebo as early as week 1. SAMe outperformed escitalopram during weeks 2, 4, and 6. Compared with escitalopram and placebo, SAMe achieved the highest response rate (45% vs 31% vs 26%) and remission rate (34% vs 23% vs 6%).
SAMe for psychiatric disorders
Aggression in people with schizophrenia is associated with a particular genetic variation in the catechol- O-methyltransferase (COMT) gene, which leads to downregulation of an enzyme that aids in neurotransmission.32 SAMe mitigated aggression in a subset of patients with schizophrenia by increasing COMT enzyme activity. Patients with chronic schizophrenia and genotype associated with reduced COMT activity were randomly assigned to receive SAMe or placebo; the SAMe group exhibited a significant reduction in aggression, as measured by the Overt Aggressiveness Scale.
The 22q11.2 deletion syndrome, involving deletion of one copy of the COMT gene, is a genetic disorder that frequently associated with mental health conditions such as psychosis, depression, and attention deficit hyperactivity disorder.33 SAMe may enhance COMT enzymatic activity, potentially alleviating mental symptoms in these patients. In a study assessing the feasibility and safety of SAMe in 12 patients with 22q11.2 deletion syndrome, SAMe treatment up to 1600 mg/day for 6 weeks appeared to be safe, well tolerated, with no serious adverse effects.34
SAMe for chronic inflammation in Alzheimer
disease
SAMe plays a crucial role in metabolic pathways that are essential for maintaining brain homeostasis. SAMe acts as a methyl donor, which affects gene expression via DNA methylation. This process leads to the production of several substances including neurotransmitters and phosphatases.35 SAMe also plays a crucial role in transsulphuration, a process that generates glutathione, the brain’s primary antioxidant. Additionally, SAMe is involved in the production of neuroregulatory polyamines, which possess anti-inflammatory characteristics and are responsible for regulating cell differentiation and proliferation. The proper functioning of these processes and subsequently of neurons relies on a sufficient supply of SAMe.
Patients with AD have significantly decreased quantities of SAMe in both their cerebrospinal fluid and post-mortem brain tissue.36 Possible causes include a lack of B12 or folate because these nutrients are necessary in producing methionine.37 Another potential cause could be the excessive use of SAMe in the synthesis of polyamines, which may be a result of a persistent inflammatory response to tau pathology. High levels of homocysteine, which occur secondary to B12 or folate deficiency, might also hinder the methylation activity of SAMe by providing negative feedback to the enzymes involved in the methionine cycle.38 The SAMe deficiency seen in the AD brain can be caused by hyperhomocysteinaemia, malnutrition, increased SAMe consumption during polyamine production in the presence of persistent neuronal inflammation, and other unidentified processes. Deficiency of SAMe is responsible for pathogenic processes that are hallmarks of AD; for example, without methylation, protein phosphatase 2A, the primary phosphatase in the brain, becomes ineffective, leading to uncontrolled tau hyperphosphorylation. The expression of genes associated with AD is a consequence of the loss of methylation in their promotors. The decrease in brain acetylcholine levels is caused by a deficiency in SAMe-mediated methylation of N-methyl nicotinamide. This deficiency often hinders the normal outflow of acetylcholine from the brain. The depletion of brain glutathione levels caused by the loss of SAMe-mediated transsulphuration impairs cerebral antioxidant action and perpetuates neuroinflammation. Irrespective of the reason, a shortage of SAMe affects several important processes that result in the pathogenesis of AD. Deficiency in SAMe can increase the expression of genes that promote the development and progression of AD, leading to elevated production of amyloid beta. It also impairs the production and retention of acetylcholine in the brain, disrupts the function of phosphatase, causes an accumulation of hyperphosphorylated tau, and obstructs the ability of glutathione to neutralise reactive oxygen species, thereby promoting a state of neuroinflammation. SAMe exhibits potential neuroprotective effects in mitigating cognitive impairment associated with brain ageing, primarily by inhibiting oxidative stress, neuroinflammation, and alpha 7 nicotinic acetylcholine receptors signalling pathways.39 Administration of SAMe in individuals with AD led to improvements in cognitive performance and severity of dementia symptoms, and the treatment was well-tolerated.
Limitations
The studies included in this review are susceptible to biases or variations in methodologies. Consequently, the generalisability of our findings may be affected by the heterogeneity of study populations and methodologies. Although the mechanisms of SAMe in terms of neurotransmitter metabolism, neuroinflammation, neuroplasticity, and gene expression regulation have been delineated to a degree, the precise molecular pathways and interactions remain incompletely understood. Further elucidation of these mechanisms is essential to optimise interventions and customise strategies to individual patients. Although SAMe supplementation is effective for depression management, comprehensive evaluation of its adverse effects, optimal dosages, treatment durations, drug interactions, and long-term safety profile is warranted to inform clinical practice. Future studies should assess the safety and tolerability of SAMe, particularly in pregnant women, children, and older adults.
Conclusion
SAMe supplementation can alleviate depression symptoms in patients with suboptimal response to standard antidepressive therapy. Current evidence suggests that SAMe, through modulating methylation processes and neurotransmitter function, may provide a complementary treatment. SAMe is a promising candidate for enhancing mental and neurological health through both standalone and adjunctive therapies.
Contributors
All authors designed the study, acquired the data, analysed the data, drafted the manuscript, and critically revised the manuscript for important intellectual content. All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding / support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Data availability
All data generated or analysed during the present study are available from the corresponding author on reasonable request.
References
- Bottiglieri T. S-Adenosyl-L-methionine (SAMe): from the bench to the bedside—molecular basis of a pleiotrophic molecule. Am J Clin Nutr 2002;76:1151S-7S.
- Blanquart C, Linot C, Cartron PF, Tomaselli D, Mai A, Bertrand P. Epigenetic metalloenzymes. Curr Med Chem 2019;26:2748-85.
- Reul JM. Making memories of stressful events: a journey along epigenetic, gene transcription, and signaling pathways. Front Psychiatry 2014;5:5.
- Artal-Martinez de Narvajas A, Gomez TS, Zhang JS, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol 2013;33:3983-93.
- Soyka M. Neurobiology of aggression and violence in schizophrenia. Schizophr Bull 2011;37:913-20.
- Mischoulon D, Fava M. Role of S-adenosyl-L-methionine in the treatment of depression: a review of the evidence. Am J Clin Nutr 2002;76:1158S-61S.
- Baldessarini RJ. Neuropharmacology of S-adenosyl-L-methionine. Am J Med 1987;83:95-103.
- Mischoulon D, Alpert JE, Arning E, Bottiglieri T, Fava M, Papakostas GI. Bioavailability of S-adenosyl methionine and impact on response in a randomized, double-blind, placebo-controlled trial in major depressive disorder. J Clin Psychiatry 2012;73:843-8.
- Fava M, Borus JS, Alpert JE, Nierenberg AA, Rosenbaum JF, Bottiglieri T. Folate, vitamin B12, and homocysteine in major depressive disorder. Am J Psychiatry 1997;154:426-8.
- Bustamante AC, Aiello AE, Galea S, et al. Glucocorticoid receptor DNA methylation, childhood maltreatment and major depression. J Affect Disord 2016;206:181-8.
- Bergink V, Gibney SM, Drexhage HA. Autoimmunity, inflammation, and psychosis: a search for peripheral markers. Biol Psychiatry 2014;75324-31.
- Spillmann M, Fava M. S-Adenosylmethionine (ademetionine) in psychiatric disorders: historical perspective and current status. CNS Drugs 1996;6:416-25.
- Gobejishvili L, Avila DV, Barker DF, et al. S-adenosylmethionine decreases lipopolysaccharide-induced phosphodiesterase 4B2 and attenuates tumor necrosis factor expression via cAMP/protein kinase A pathway. J Pharmacol Exp Ther 2011;337433-43.
- Ara AI, Xia M, Ramani K, Mato JM, Lu SC. S-adenosylmethionine inhibits lipopolysaccharide-induced gene expression via modulation of histone methylation. Hepatology 2008;47:1655-66.
- Saunderson EA, Spiers H, Mifsud KR, et al. Stress-induced gene expression and behavior are controlled by DNA methylation and methyl donor availability in the dentate gyrus. Proc Natl Acad Sci U S A 2016;113:4830-5.
- Pfalzer AC, Choi SW, Tammen SA, et al. S-adenosylmethionine mediates inhibition of inflammatory response and changes in DNA methylation in human macrophages. Physiol Genomics 2014;46:617- 23.
- Cleare A, Pariante CM, Young AH, et al. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2008 British Association for Psychopharmacology guidelines. J Psychopharmacol 2015;29:459-525.
- Gao J, Cahill CM, Huang X, et al. S-adenosyl methionine and transmethylation pathways in neuropsychiatric diseases throughout life. Neurotherapeutics 2018;15:156-75.
- Papakostas GI, Mischoulon D, Shyu I, Alpert JE, Fava M. S- adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder: a double-blind, randomized clinical trial. Am J Psychiatry 2010;167:942-8.
- Benelli A, Filaferro M, Bertolini A, Genedani S. Influence of S- adenosyl-L-methionine on chronic mild stress-induced anhedonia in castrated rats. Br J Pharmacol 1999;127:645-54.
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003;33 Suppl:245-54.
- Polis B, Karasik D, Samson AO. Alzheimer’s disease as a chronic maladaptive polyamine stress response. Aging (Albany NY) 2021;13:10770-95.
- Otero-Losada ME, Rubio MC. Acute changes in 5-HT metabolism after S-adenosyl-L-methionine administration. Gen Pharmacol 1989;20:403-6.
- Lu SC. S-Adenosylmethionine. Int J Biochem Cell Biol 2000;32:391- 5.
- Bellido I, Gomez-Luque A, Plaza A, Rius F, Ortiz P, Sanchez de la Cuesta F. S-adenosyl-l-methionine prevents 5-HT1A receptors up- regulation induced by acute imipramine in the frontal cortex of the rat. Neurosci Lett 2002;321:110-4.
- Sarris J, Papakostas GI, Vitolo O, Fava M, Mischoulon D. S-adenosyl methionine (SAMe) versus escitalopram and placebo in major depression RCT: efficacy and effects of histamine and carnitine as moderators of response. J Affect Disord 2014;164:76-81.
- Bottiglieri T, Arning E, Wasek B, Nunbhakdi-Craig V, Sontag JM, Sontag E. Acute administration of L-DOPA induces changes in methylation metabolites, reduced protein phosphatase 2A methylation, and hyperphosphorylation of Tau protein in mouse brain. J Neurosci 2012;32:9173-81.
- De Berardis D, Orsolini L, Serroni N, et al. A comprehensive review on the efficacy of S-Adenosyl-L-methionine in major depressive disorder. CNS Neurol Disord Drug Targets 2016;15:35-44.
- Galizia I, Oldani L, Macritchie K, et al. S-adenosyl methionine (SAMe) for depression in adults. Cochrane Database Syst Rev 2016;10:CD011286.
- Sharma A, Gerbarg P, Bottiglieri T, et al. S-Adenosylmethionine (SAMe) for neuropsychiatric disorders: a clinician-oriented review of research. J Clin Psychiatry 2017;78:e656-67.
- Kagan BL, Sultzer DL, Rosenlicht N, Gerner RH. Oral S- adenosylmethionine in depression: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry 1990;147:591-5.
- Strous RD, Nolan KA, Lapidus R, Diaz L, Saito T, Lachman HM. Aggressive behavior in schizophrenia is associated with the low enzyme activity COMT polymorphism: a replication study. Am J Med Genet B Neuropsychiatr Genet 2003;120B:29-34.
- Tang KL, Antshel KM, Fremont WP, Kates WR. Behavioral and psychiatric phenotypes in 22q11.2 deletion syndrome. J Dev Behav Pediatr 2015;36:639-50.
- Green T, Steingart L, Frisch A, Zarchi O, Weizman A, Gothelf D. The feasibility and safety of S-adenosyl-L-methionine (SAMe) for the treatment of neuropsychiatric symptoms in 22q11.2 deletion syndrome: a double-blind placebo-controlled trial. J Neural Transm (Vienna) 2012;119:1417-22.
- Otero Losada ME, Rubio MC. Acute effects of S-adenosyl-L- methionine on catecholaminergic central function. Eur J Pharmacol 1989;163:353-6.
- McGrowder DA, Miller F, Vaz K, et al. Cerebrospinal fluid biomarkers of Alzheimer’s disease: current evidence and future perspectives. Brain Sci 2021;11:1-56.
- Hama Y, Hamano T, Shirafuji N, et al. Influences of folate supplementation on homocysteine and cognition in patients with folate deficiency and cognitive impairment. Nutrients 2020;12:3138.
- Mihara A, Ohara T, Hata J, et al. Association of serum s- adenosylmethionine, s-adenosylhomocysteine, and their ratio with the risk of dementia and death in a community. Sci Rep 2022;12:12427.
- Zhang Y, Ma R, Deng Q, et al. S-adenosylmethionine improves cognitive impairment in D-galactose-induced brain aging by inhibiting oxidative stress and neuroinflammation. J Chem Neuroanat 2023;128:102232.