
Hong Kong J Psychiatry 2006;16:150-3
Original Article
Bipolar Mood Disorder Secondary to Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-likeEpisodes (MELAS): a Case Report
因MELAS而誘發的一個雙極情感性疾病個案
K Kannan, SMK Tan, SK Pillai, D Sinniah, AA Raymond, AH Hamzaini, SF Loh, WS Ismail, V Swami, N Ruzyanie
Dr Kumaraswami Kannan, Hospital Mesra Bukit Padang, Kota Kinabalu, Malaysia.
Dr Susan MK Tan, Department of Psychiatry, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia.
Dr Subash K Pillai, Universiti Malaya Medical Centre, Kuala Lumpur, Malaysia.
Dr Dhachayani Sinniah, Department of Psychiatry, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia.
Dr AA Raymond, Division of Neurology, Department of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia.
Dr AH Hamzaini, Department of Radiology, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia.
Dr SF Loh, Department of Psychiatry, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia.
Dr Wan S Ismail, Department of Psychiatry, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia.
Dr Viren Swami, Department of Public Health, University of Liverpool, Liverpool, United Kingdom.
Dr Nik Ruzyanie, Department of Psychiatry, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia.
Address correspondence to: Dr Kumaraswami Kannan, Hospital Mesra Bukit Padang, Peti Surat 11342, 88815 Kota Kinabalu, Sabah, Malaysia.
E-mail: drks10@hotmail.com
Submitted: 7 November 2006; Accepted: 13 February 2007
Abstract
Psychiatric symptoms in patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome are rarely reported. We report a case of bipolar mood disorder, secondary to MELAS, in an adolescent female patient. To our knowledge, this is the first such report in the literature.
Key words:Bipolar disorder; MELAS syndrome
摘要
文獻很少記載有關粒線體腦肌肉病變,乳酸毒症和類中風發作綜合症患者(MELAS ) 的精神症狀。我們報告一位因MELAS而誘發雙極情威性疾病的個案。據讀我們所知,這是首個有關的病例報告。
關鍵詞:雙極情感性疾病、粒線體腦肌肉病變乳酸毒症和類中風發作綜合症
Introduction
Recent research has documented several encephalo- myopathies related to mitochondrial dysfunction, one of the most important being mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome.1 The clinical expression of MELAS is highly variable.2 In most cases (80%), however, the enzymatic defect in MELAS is a complex I deficiency associated with a point mutation at np3243 in the tRNALeu(UUR) region.3
Mental deterioration usually progresses after repeated episodic attacks, and may lead to psychiatric abnormalities including schizophrenia and dementia.4 Here, we report a case of bipolar mood disorder, secondary to MELAS, in a female patient.
Case report
In December 2004, a 15-year-old Malay girl was brought by her parents to the accident and emergency department with a sudden onset of right-sided weakness. Prior to this episode she had been a healthy child and had suffered no significant medical illnesses in the past. A brain magnetic resonance imaging (MRI) showed multifocal enhancing lesions in both basal ganglia, both internal capsules, and the brainstem. Over the next few weeks her condition worsened and she developed expressive aphasia. She also became depressed and was started on an anti-depressant (citalopram, 20 mg). Following discharge, she continued taking the anti- depressant for 90 days, before defaulting and resorting to traditional medicine (prayers, baths, and holy water).
By March 2005, she had regained most of her motor functions but developed manic symptoms including inflated self-esteem, pressured speech with flight of ideas, and an increase in motor activity, which led to marked distress and impairment in her daily functioning. In December 2005, she was referred to our adolescent psychiatric unit for progressive worsening of her symptoms. She was euphoric, talked incessantly about a single topic, showed disinhibition and was excessively energetic but, showed no signs of abulia (absence of urges, desires, or interests). Following psychiatric tests, she was diagnosed Bipolar I mood disorder in manic phase.
A repeat MRI of the brain showed a reduction in the size of the lesions in the periventricular region (bilaterally), right corona radiata, body of the corpus callosum (right), basal ganglia (bilaterally), left thalamus, and midbrain. These lesions were relatively hypointense on T1-weighted imaging and non-enhancing post-contrast. Some of the periventricular lesions were oval in shape and located perpendicular to the ventricles. There was no evidence of any juxtacortical lesions or abnormal signal intensity within the cerebellum and the imaged portion of the orbit. The MRI features were suggestive of a white matter demyelinating disease. Differential diagnoses included multiple sclerosis, ischaemic microvascular disease, mitochondrial disease, systemic lupus erythematosus (SLE), and sarcoidosis. A panel of neurologists made a provisional diagnosis of MELAS, after taking into account the presenting history coupled with the MRI findings.
In December 2005, the patient began receiving twice- daily doses of the anti-psychotic quetiapine (300 mg) to control her agitation and delusions and sodium valproate (Epilim, 600 mg twice daily; Sanofi-Synthelabo, Newcastle- upon-Tyne, UK) as a mood stabiliser. She was also given a course of oral benzodiazepine (lorazepam), which was gradually tapered off over 2 weeks. On assessment by a neurologist, aspirin (150 mg daily) was initiated. No steroids nor other mania-inducing drugs were ever started. To further assist her recovery, speech therapy and physiotherapy were provided and eventually led to a marked improvement in her symptoms.
In January 2006, the patient was readmitted following a sudden-onset hemiparesis lasting 2 hours. She was diagnosed as having had a transient ischaemic attack. During this admission, the on-call psychiatric team reassessed her and found that she was irritable, over-familiar, energetic, and mildly labile. Her dose of quetiapine was increased by 100 mg, while that of sodium valproate was maintained.
Over the next 6 months, the patient regained her motor functions and showed a gradual improvement in her manic symptoms. In April 2006, however, follow- up psychiatric tests showed that the patient’s IQ, as measured on the Weschler Intelligence Scale for Children, had deteriorated and was now exceptionally low on both verbal and performance scales. Other neuropsychiatric tests showed no impairment in her long-term memory, although there was a slight interference with short-term memory. There was also some indication of frontal lobe dysfunction, including trouble acquiring, retaining and retrieving verbal information, difficulty with visual scanning and coordination, and poor organisation strategies. The temporal relationship between her manic symptoms and the onset of the medical conditions is shown in the Table (Figs 1, 2).

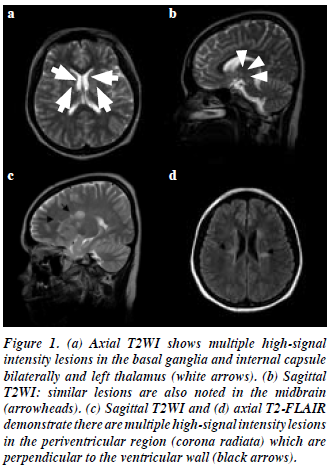

Discussion
‘ MELAS ’ stands for mitochon d r i almyopathy, encephalopathy, lactic acidosis, and symptoms of stroke and was first described in 1984 by Pavlakis et al.5 It is caused by a genetic defect in the mitochondria; most MELAS patients have a heteroplasmic A-to-G point mutation in the dihydrouridine loop of the transfer RNA (tRNA)Leu(UUR) gene at base pair (bp) 3243 (i.e. 3243 AG→mutation). The heteroplasmy in MELAS varies from patient to patient, reflecting variable segregation in the ovum.6
A study of the epidemiology of mitochondrial disorders found a prevalence of more than 10.2 per 100,000 for the 3243A→G mutation in the adult Finnish population.7
If all first-degree maternal relatives of a verified mutation carrier also harbour the mutation, the prevalence may be more than 16.3 per 100,000, making mitochondrial disorders potentially one of the major neurogenetic diseases in adults. A study of a northern English adult population found this mutation in approximately 1 per 13,000.8
The MELAS disorder–associated human mitochon- drial tRNALeu(UUR) mutation causes an aminoacylation deficiency and a concomitant defect in translation initiation. It involves most organ systems including the central nervous system, skeletal muscle, eyes, cardiac muscle, and, more rarely, the gastro-intestinal and renal systems. Symptoms of MELAS include clinical stroke, seizures, lactic acidosis, and exercise intolerance. Additional clinical manifestations include dementia, limb weakness, short stature, and recurrent migraine-like headaches. To date, psychiatric symptoms have rarely been reported in patients with MELAS.9
The stroke-like episodes provide an ongoing insult to the brain that precipitates and perpetuates mental deterioration, which in our patient initially presented as depression, hemiparesis and aphasia, and later as mania.
The diagnosis of MELAS is usually made on clinical grounds but confirmation requires a muscle or brain biopsy. If the patient does indeed suffer from MELAS, a muscle biopsy, for example, shows ragged red muscle fibres suggestive of a mitochondrial disorder. In this case, no biopsy was done as the parents did not consider it necessary and wanted to spare their child undue suffering.
Suzuki et al10 described 1 patient with MELAS who had schizophrenia-like symptoms including auditory hallucinations and delusions of persecution. Thormeer et al4 reported on a female patient with MELAS in whom psychiatric symptoms included delusions of reference, paranoia, and auditory hallucinations. In our patient the diagnosis of MELAS was established by MRI evidence of multiple areas of increased signal on T2WI and fluid- attenuated inversion recovery (FLAIR) prior to the appearance of her psychiatric symptoms.
The neurological abnormalities described in this case, such as expressive aphasia and hemiparesis, are generally in agreement with those reported by other investigators4,10 but this report is perhaps unique in describing additional psychiatric symptomatology, namely bipolar mood disorder. Nevertheless, manic symptoms are not inconsistent with MELAS, as indicated by the link between manic symptoms and the type of brain lesions seen in our patient.11 It is possible that the mood disorder exhibited by our patient was caused by the site of the brain lesions rather than through a direct link between MELAS and bipolar moods.
The short-term prognosis for this patient is good considering the high level of family support, good response to medication, and the lack of any permanent disability or organ complications. She eventually returned to school and participated in class activities, although she was not able to sit for any examinations. Her long-term prognosis, however, is poor: MELAS has a chronic course and is progressive in nature. Other poor prognostic factors include the marked deterioration in her intellectual ability, the early onset of the disease, and the strength of the familial belief in alternative medicines.
Although MELAS is a rare disease, this report highlights the importance of considering mitochondrial encephalomyopathy as a diagnosis in complex psychiatric presentations.
References
- Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M, et al. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol 1992;31:391-8.
- Crimmins D, Morris JG, Walker GL, Sue CM, Byrne E, Stevens S, et al. Mitochondrial encephalomyopathy: variable clinical expression within a single kindred. J Neurol Neurosurg Psychiatry 1993;56:900- 5.
- Shapira Y. Clinical aspects of mitochondrial encephalomyopathies. Int Pediatr 1993;8:225-32.
- Thomeer EC, Verhoeven WM, van de Vlasakker CJ, Klompenhouwer JL. Psychiatric symptoms in MELAS: a case report. J Neurol Neurosurg Psychiatry 1998;64:692-3.
- Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol 1984;16:481-8.
- Cardaioli E, Fabrizi GM, Grieco GS, Dotti MT, Federico A. Heteroplasmy of the A3243G transition of mitochondrial tRNALeu(UUR) in a MELAS case and in a 25-week-old miscarried fetus. J Neurol 2000;247:885-7.
- Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Kärppä M, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Hum Genet 1998;63:447-54.
- Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders—past, present and future. Biochim Biophys Acta 2004;1659:115-20.
- MELAS syndrome. e-medicine from WebMD website: http://www.emedicine.com/ped/topic1406.htm. Accessed 15 Aug 07.
- Suzuki T, Koizumi J, Shiraishi H, Ishikawa N, Ofuku K, Sasaki M, et al. Mitochondrial encephalomyopathy (MELAS) with mental disorder CT, MRI and SPECT findings. Neuroradiology 1990;32:74-6.
- Starkstein SE, Mayberg HS, Barthier ML, Fedoroff P, Price TR, Dannals RF. Mania after brain injury: neuroradiological and metabolic findings. Ann Neurol 1990;27:652-9.
